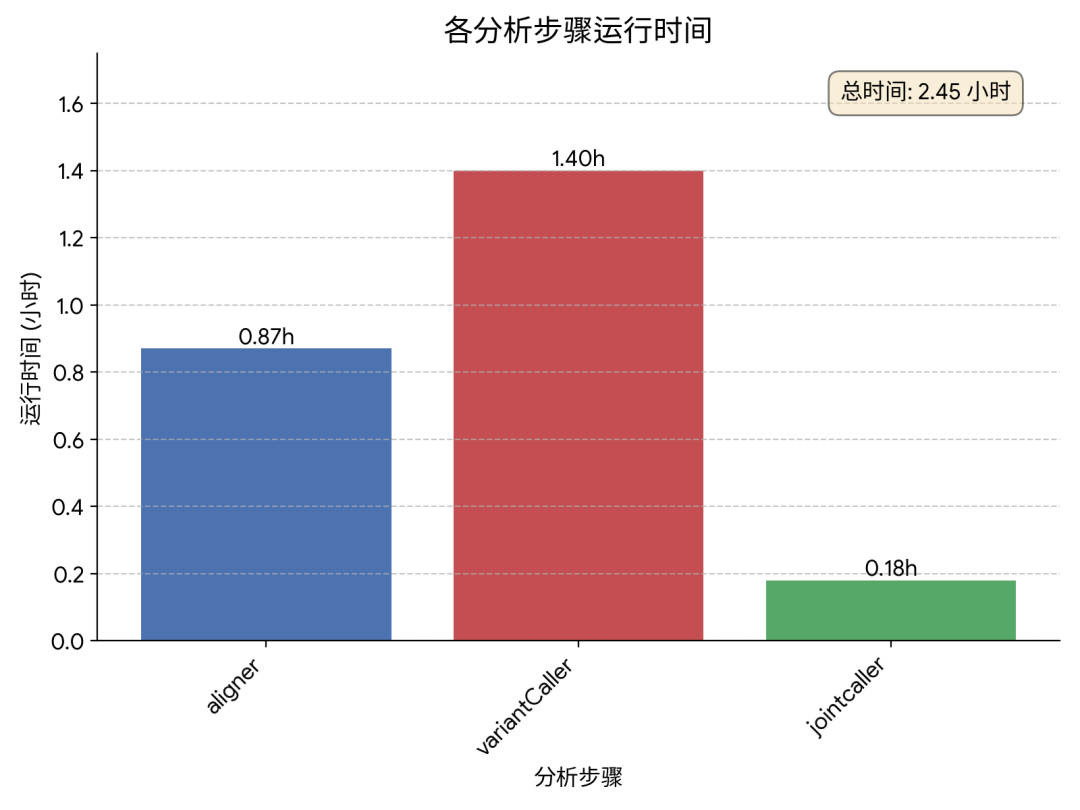
1 大型队列研究
大型队列研究往往通过对群体样本做WGS/WES(Whole Genome/Exome Sequencing, 全基因组/外显子组测序)测序来展开研究。其核心分析流程主要包括单样本水平上的变异检测、联合变异检测和下游分析。
其中单样本变异检测流程一般包括数据质控、序列比对、标记重复、碱基质量值重校正、变异检测等步骤。DCS tools 遵循原始算法,对算法实现进行了性能优化,优化后的分析流程比BWA-GATK快16倍。联合变异检测部分DCS Tools提供jointcaller这一基于Spark分布式计算框架的工具,能够高效处理百万级样本输入。
1.1 准备工作
WGS分析流程的输入文件如下:
-
序列比对工具需要的文件;
索引文件 描述 species.fa 参考基因组FASTA文件 species.fa.fai 用于快速定位和随机访问FASTA文件的序列区域 species.dict 索引作用,为后续处理流程提供参考序列的快速查找表 species.fa.* 比对索引文件 通过aligner工具构建比对索引,索引文件放置在FASTA同级目录,比对索引构建命令是:
dcs aligner --threads <INT> --build-index <FASTA>
-
FASTQ测序数据,支持gz格式;
-
单核苷酸多态性数据库(dbSNP)数据和已知变异位点文件,以VCF格式提供;
1.2 序列比对和标记重复
将序列比对到参考基因组上确定每条序列的位置,为进一步检测变异提供基础;标记重复消除PCR扩增产生的重复序列,降低假阳性提高变异检测准确性,这些步骤由软件alinger完成。而且aligner 还集成了数据质控功能,可以在序列比对之前对序列进行过滤和处理,从而降低错误率,避免后续分析受到噪音数据的干扰。
dcs aligner --threads THREADNUM \
--aln-index INDEX \
--fq1 FASTQ1 \
--fq2 FASTQ2 \
--read-group RGROUP \
--bam-out BAMFILE \
--fastq-qc-dir QCDIR
该命令需要以下输入:
1. THREADNUM: 线程数量,建议该值不要超过CPU数量;
2. INDEX: 参考基因组FASTA文件路径,在FASTA同级目录下存放比对软件构建的索引文件;
3. FASTQ1: PE测序的FASTQ1文件路径;
4. FASTQ2: PE 测试的FASTQ2文件路径;
5. RGROUP: 格式是 “@RG\tID:GROUP_NAME\tSM:SAMPLE_NAME\tPL:PLATFORM“, 其中GROUP_NAME 是read group标识符,SAMPLE_NAME表示样品名称,PLATFORM表示测序平台;
6. BAMFILE: 经过标记重复后的BMA文件;
7. QCDIR: 质控报告的输出位置;
1.3 碱基质量值矫正
由于存在系统误差,序列碱基质量值并不总是完全准确,需要对序列的碱基质量值重校正。该步骤可以纠正系统偏差生成更精确的质量分数,提高变异检测准确性,减少假阳性和假阴性。统一质量评估标准符合GATK推荐的最佳实践,该步骤由软件bqsr完成。
dcs bqsr --threads THREADNUM \
--in-bam BAMFILE \
--reference FASTA \
--known-sites KNOWN_SITES \
--recal-table RECAL_TABLE
该命令需要以下输入:
1. THREADNUM: 线程数量,建议该值不要超过CPU核数;
2. BAMFILE: 输入文件,序列比对和标记重复后生成的BAM文件;
3. FASTA: 参考基因组FASTA文件路径,确保使用参考基因组和序列比对阶段使用的相同;
4. KNOWN_SITES: 单核苷酸多态性数据库数据和已知变异位点文件;
5. RECAL_TABLE: 输出文件,保存用来矫正碱基质量值的信息
1.4 变异检测
检测SNP和INDEL变异,生成gvcf文件,同时生成bam的统计信息,统计文件与GVCF同目录,名字为bamStat.txt。该步骤由软件variantcaller完成
dcs variantCaller -I INPUT_BAM \
-O OUTPUT_GVCF \
-R REFERENCE \
--recal-table RECAL_TABLE
-t THREADUM
该命令需要以下输入:
1. INPUT_BAM:2.1.2输出的BAM文件(排序标记重复后)。
2. OUTPUT_GVCF:输出gvcf文件。
3. REFERENCE:参考基因组FASTA文件。
4. RECAL_TABLE:输入文件,包含矫正碱基质量值所需要的信息,由2.1.3步骤生成。
5. THREADNUM:线程数量,建议该值不要超过CPU核数。
对于单个样本,如果仅仅需要获取包含变异记录的vcf文件,可以使用genotyper软件。
dcs genotyper -I INPUT_GVCF \
-O OUTPUT_VCF \
-s QUAL \
-t THREDNUM
该命令需要以下输入:
1. INPUT_GVCF:来自于variantcaller的GVCF文件。
2. OUTPUT_VCF:输出VCF文件。
3. QUAL:变异质量值阈值,大质量值大于或等于QUAL的记录将被输出。
4. THREADNUM:线程数量,建议该值不要超过CPU核数。
1.5 报告生成
可以根据每个样本的统计文件进行整合,生成可视化HTML报告文件
dcs report --sample SAMPLE_NAME \
--filter QCDIR \
--bam_stat BAMStat \
--vcf_stat VCFStat \
--output OUTPUT_DIR
该命令需要以下输入:
1. SAMPLE_NAME:样本命名。
2. QCDIR:aligner生成的质控统计报告文件夹。
3. BAMStat:variantCaller结果目录下的bamStat.txt。
4. VCFStat:vcf统计结果,默认在工作目录下的vcfStat.txt。
5. OUTPUT_DIR: 报告输出目录。
1.6 联合变异检测
将多个样本的GVCF作为输入,进行联合变异检测,生成合并多个样本的变异并重新计算样本基因型。DCS Tools的jointcaller是一个基于spark框架的程序,可以在多台机器上以单机模式运行,示例如下:
# 设置JAVA_HOME为jdk8
export JAVA_HOME=/path_to/jdk8
# 将spark bin加入到PATH,用于调用spark-submit
export PATH=/path_to/spark/bin/:$PATH
dcs jointcaller \
-i GVCF_LIST \
-r REFERENCE \
-o OUTDIR \
-j JOBS \
--memory MEMORY \
-l REGION
DPGT依赖java8,请设置JAVA_HOME=/path_to/jdk8。spark推荐2.4.5及以上的版本,spark 2.4.5下载路径:https://archive.apache.org/dist/spark/spark-2.4.5/spark-2.4.5-bin-hadoop2.6.tgz
该命令需要如下输入:
GVCF_LIST:输入的gvcf列表,是一个文本文件,每行一个gvcf路径。注意gvcf需要有匹配的tbi索引文件,程序默认通过gvcf路径+.tbi查找gvcf相匹配的tbi索引文件。
REFERENCE:参考基因组fasta文件路径,注意这个fasta文件需要与生成gvcf是使用的fasta文件一致。程序同时需要与fasta文件匹配的.dict和.fai文件,例如fasta文件名是hg38.fasta,那么程序也需要读取hg38.dict, hg38.fasta.fai。
OUTDIR:输出文件夹,结果vcf路径是OUTDIR/result.*.vcf.gz。
JOBS:并行的任务数目,使用的线程数目是JOBS个,JOBS是一个整型值,范围是1到机器的线程数目。
MEMORY:java最大堆内存大小,内存的单位是"g"或"m"。内存大小和JOBS值和样本量存在关联,对于1000样本,推荐每个job分配2g内存,对于2000-5000样本,推荐每个job分配4g内存,对于5000-100000样本,推荐每个job分配6g内存,对于100000-500000样本,推荐每个job分配8g内存。
REGION:目标区域。支持输入字符串和一个bed路径,区域字符串格式与bcftools定义的格式一致,例如:"chr20:1000-2000", "chr20:1000-", "chr20",如果需要指定多个区域,请指定多个-l参数,例如指定处理chr1和chr2:1000-2000可以这样做:-l chr1 -l chr2:1000-2000
1.7 WES
如果队列研究采用的是WES或者其它panel方案,那么上述过程中有两步需要调整
1. variantCaller步骤添加区间信息(-L参数)。参考命令如下:
dcs variantCaller -I INPUT_BAM \
-O OUTPUT_GVCF \
-R REFERENCE \
--recal-table RECAL_TABLE
-L INTERVAL_FILE
-t THREADUM
INTERVAL_FILE:记录WES区间的bed文件,按照惯例其中每行格式如下
chr<tab>start<tab>end
其中start和end位置是0-based表示,属于左闭右开的区间。
2. BAM统计需要单独执行,使用DCS Tools的util模块中的bam-stat功能来完成。
2 遗传病诊断
相比大型队列研究,该场景下一般不需要做联合变异检测,除了1.6步骤,其它步骤相同。即每个样本执行如下分析步骤:
1. 序列比对和标记重复(aligner)
2. 碱基质量值矫正(bqsr)
3. 变异检测(包括variantCaller和genotyper)
4. 统计报告生成(report)
后续步骤一般可使用其它工具来做变异注释和致病性分析等。
3 肿瘤诊断
Mutect2可以检测体细胞snp和InDel。该工具可以在三种场景模式下运行:(i) 肿瘤-正常模式,其中肿瘤样本与正常样本进行匹配分析;(ii) 仅肿瘤模式,其中对单个样本的比对数据进行分析;以及 (iii) 线粒体模式,该模式需要高深度、高灵敏度的基因分型。DCS Tools的Mutect2工具是GATK Mutect2(v4.6.2.0)的加速版本,增加了并行处理功能,该功能通过设置--nthread参数来开启,在 32 线程下可以获得约 8 倍左右的速度提升(实际情况会受机器性能影响)。
我们以场景(i)为例说明DCS Tools Mutect2的使用方法。参考1.2-1.3步骤,得到了正常样本和肿瘤样本的BAM文件后,可以按照下面命令进行体细胞变异检测:
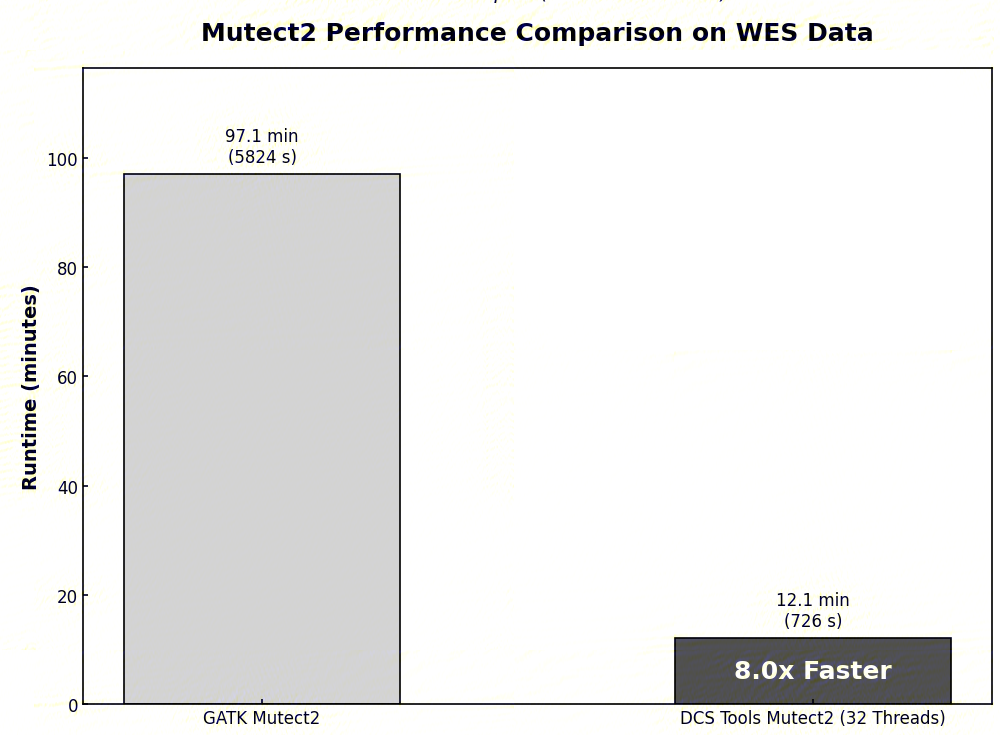
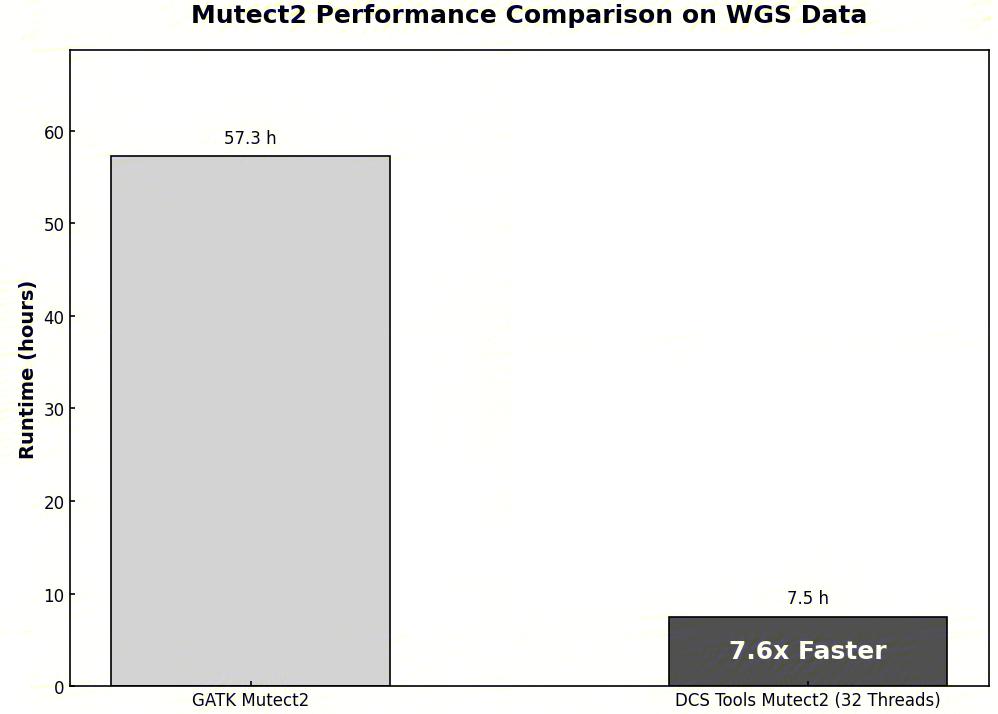
使用命令如下:
$DCS_HOME/bin/dcs Mutect2 -R <ref.fasta> \
-I <tumor.bam> \
-I <normal.bam> \
-normal normal \
-O <somatic.vcf.gz> \
--nthread 32
其它场景的命令可以参考如下:
dcs Mutect2 --java-options "<Xmx等JVM参数>" --nthreads <线程数> [其它GATK参数]
”其它GATK参数“可以参考GATK Mutect2的文档(https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2)。
4 动植物基因组学
动植物基因组30X数据测试显示,单样本变异检测大部分在2小时内完成分析,与GATK的一致性SNP达到99.98%以上,Indel达99%以上,并且支持多倍体分析(在variantCaller步骤设置ploidy参数)。分析步骤与上面大型队列类似,其中碱基质量值矫正步骤为可选步骤(如果该物种有相应数据库,建议开启该步骤)。